

(Noticias de Nanowerk) Los anticuerpos, pequeñas proteínas producidas por el sistema inmunitario, pueden adherirse a partes específicas de un virus para neutralizarlo. Mientras los científicos continúan luchando contra el SARS-CoV-2, el virus que causa el covid-19, una posible arma es un anticuerpo sintético que se une a las proteínas de punta del virus para evitar que el virus ingrese a una célula humana.
Para desarrollar un anticuerpo sintético exitoso, los investigadores deben comprender exactamente cómo sucederá esa unión. Las proteínas, con estructuras 3D grumosas que contienen muchos pliegues, pueden unirse en millones de combinaciones, por lo que encontrar el complejo proteico correcto entre casi innumerables candidatos requiere mucho tiempo.
Para agilizar el proceso, los investigadores del MIT crearon un modelo de aprendizaje automático que puede predecir directamente el complejo que se formará cuando dos proteínas se unan. Su técnica es entre 80 y 500 veces más rápida que los métodos de software más avanzados y, a menudo, predice estructuras de proteínas que están más cerca de las estructuras reales que se han observado experimentalmente.
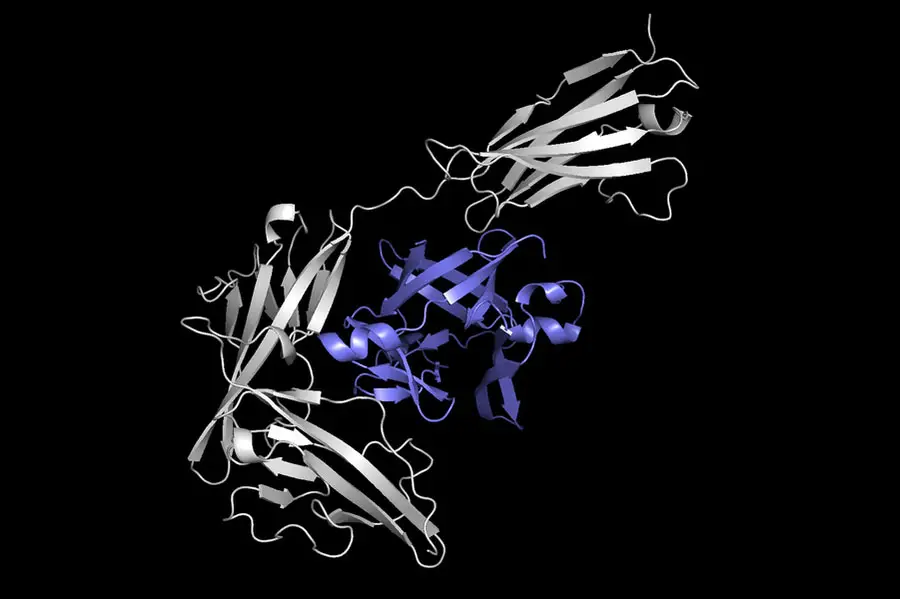 Esta imagen muestra una proteína (en gris) acoplada con otra proteína (en púrpura) para formar un complejo proteico. Equidock, el sistema de aprendizaje automático que desarrollaron los investigadores, puede predecir directamente un complejo proteico como este en cuestión de segundos. (Imagen: Cortesía de los investigadores)
Esta imagen muestra una proteína (en gris) acoplada con otra proteína (en púrpura) para formar un complejo proteico. Equidock, el sistema de aprendizaje automático que desarrollaron los investigadores, puede predecir directamente un complejo proteico como este en cuestión de segundos. (Imagen: Cortesía de los investigadores)
Esta técnica podría ayudar a los científicos a comprender mejor algunos procesos biológicos que involucran interacciones de proteínas, como la replicación y reparación del ADN; también podría acelerar el proceso de desarrollo de nuevos medicamentos.
“El aprendizaje profundo es muy bueno para capturar interacciones entre diferentes proteínas que, de otro modo, serían difíciles de escribir experimentalmente para los químicos o biólogos. Algunas de estas interacciones son muy complicadas y la gente no ha encontrado buenas maneras de expresarlas. Este modelo de aprendizaje profundo puede aprender este tipo de interacciones a partir de los datos”, dice Octavian-Eugen Ganea, posdoctorado en el Laboratorio de Ciencias de la Computación e Inteligencia Artificial (CSAIL) del MIT y coautor principal del artículo («Independent SE(3) -Modelos equivalentes para el acoplamiento de proteínas rígidas de extremo a extremo»).
El coautor principal de Ganea es Xinyuan Huang, estudiante graduado en ETH Zurich. Los coautores del MIT incluyen a Regina Barzilay, Profesora Distinguida de IA y Salud de la Escuela de Ingeniería en CSAIL, y Tommi Jaakkola, Profesor de Ingeniería Eléctrica Thomas Siebel en CSAIL y miembro del Instituto de Datos, Sistemas y Sociedad. La investigación será presentada en la Conferencia Internacional sobre Representaciones de Aprendizaje.
unión a proteínas
El modelo que desarrollaron los investigadores, llamado Equidock, se enfoca en el acoplamiento de cuerpos rígidos, que ocurre cuando dos proteínas se unen al rotar o trasladarse en el espacio 3D, pero sus formas no se contraen ni se doblan.
El modelo toma las estructuras 3D de dos proteínas y las convierte en gráficos 3D que pueden ser procesados por la red neuronal. Las proteínas se forman a partir de cadenas de aminoácidos, y cada uno de esos aminoácidos está representado por un nodo en el gráfico.
Los investigadores incorporaron conocimientos geométricos en el modelo, por lo que comprende cómo pueden cambiar los objetos si se giran o trasladan en el espacio 3D. El modelo también tiene conocimiento matemático incorporado que asegura que las proteínas siempre se adhieran de la misma manera, sin importar dónde se encuentren en el espacio 3D. Así es como las proteínas se acoplan en el cuerpo humano.
Usando esta información, el sistema de aprendizaje automático identifica átomos de las dos proteínas que tienen más probabilidades de interactuar y formar reacciones químicas, conocidas como puntos de unión. Luego usa estos puntos para juntar las dos proteínas en un complejo.
“Si podemos comprender a partir de las proteínas qué partes individuales es probable que sean estos puntos de unión, entonces eso capturará toda la información que necesitamos para colocar las dos proteínas juntas. Suponiendo que podamos encontrar estos dos conjuntos de puntos, entonces podemos descubrir cómo rotar y traducir las proteínas para que un conjunto coincida con el otro conjunto”, explica Ganea.
Uno de los mayores desafíos de construir este modelo fue superar la falta de datos de entrenamiento. Debido a que existen tan pocos datos experimentales en 3D para las proteínas, fue especialmente importante incorporar el conocimiento geométrico en Equidock, dice Ganea. Sin esas restricciones geométricas, el modelo podría detectar correlaciones falsas en el conjunto de datos.
Segundos vs horas
Una vez que se entrenó el modelo, los investigadores lo compararon con cuatro métodos de software. Equidock es capaz de predecir el complejo proteico final después de solo uno a cinco segundos. Todas las líneas de base tomaron mucho más tiempo, entre 10 minutos y una hora o más.
En las medidas de calidad, que calculan en qué medida el complejo proteico pronosticado coincide con el complejo proteico real, Equidock a menudo fue comparable con las líneas de base, pero a veces las superó.
“Todavía estamos rezagados con respecto a una de las líneas de base. Nuestro método aún se puede mejorar y aún puede ser útil. Podría usarse en una proyección virtual muy grande en la que queremos comprender cómo miles de proteínas pueden interactuar y formar complejos. Nuestro método podría usarse para generar un conjunto inicial de candidatos muy rápido, y luego estos podrían ajustarse con algunos de los métodos tradicionales más precisos, pero más lentos”, dice.
Además de usar este método con modelos tradicionales, el equipo quiere incorporar interacciones atómicas específicas en Equidock para poder hacer predicciones más precisas. Por ejemplo, a veces los átomos de las proteínas se unirán a través de interacciones hidrofóbicas, que involucran moléculas de agua.
Su técnica también podría aplicarse al desarrollo de pequeñas moléculas similares a las drogas, dice Ganea. Estas moléculas se unen a las superficies de las proteínas de maneras específicas, por lo que determinar rápidamente cómo se produce esa unión podría acortar el tiempo de desarrollo del fármaco.
En el futuro, planean mejorar Equidock para que pueda hacer predicciones sobre el acoplamiento flexible de proteínas. El mayor obstáculo es la falta de datos para el entrenamiento, por lo que Ganea y sus colegas están trabajando para generar datos sintéticos que podrían usar para mejorar el modelo.