


AlphaFold 2 de DeepMind es capaz de predecir la forma tridimensional en la que se plegarán cadenas específicas de aminoácidos.
Crédito: DeepMind
DeepMind, con sede en Londres, es conocida por utilizar el aprendizaje profundo para vencer al mejor jugador de Go de la humanidad. En diciembre, sin embargo, la organización reveló su destreza contra la madre naturaleza al predecir la forma tridimensional en la que se plegarán cadenas específicas de aminoácidos.
Reconociendo la importancia biológica y médica de la pregunta plegable, los investigadores tienen un proyecto de larga data para comparar diferentes algoritmos. Esta «Evaluación crítica de la predicción de la estructura de las proteínas» bienal, o CASP, ha documentado un progreso constante durante los últimos 26 años. La entrada de DeepMind, AlphaFold 2, voló el campo.
«Casi lo entendieron», dijo Andrei Lupas del Instituto Max Planck de Biología del Desarrollo en Tübingen, Alemania, quien se ofrece como voluntario como «asesor» de CASP. «Fue el tipo de avance que cambia el juego por completo».
Las proteínas son moléculas largas que encadenan decenas o cientos de los 20 aminoácidos. Su orden exacto está especificado por la información genética en el ADN, que ahora se determina fácil y económicamente. Sin embargo, para comprender la función de las proteínas, los investigadores necesitan saber cómo esta cadena se pliega en una máquina molecular compacta, eligiendo una configuración entre una gran cantidad de posibilidades (históricamente estimada como 10300).
La estructura de la proteína se derivó históricamente de la dispersión de rayos X, que requiere una preparación minuciosa de cristales purificados grandes o de resonancia magnética nuclear. Más recientemente, el campo se ha beneficiado de la caracterización con microscopio electrónico de muestras más pequeñas congeladas instantáneamente.
Estos experimentos han revelado importantes principios organizativos, como atracciones entre varios tipos de aminoácidos, así como motivos estructurales recurrentes como hélices y láminas. Sin embargo, predecir qué configuración general tiene la energía más baja solo a partir de la secuencia, sigue siendo una tarea computacional abrumadora.
Evaluación cegada
Al principio, existía la preocupación de que los investigadores pudieran estar modificando sus programas, quizás sin querer, para que se ajustaran mejor a la estructura proteica medida. Iniciado en 1994, CASP evitó este problema al anunciar las secuencias de proteínas pero reteniendo la estructura hasta después de que se terminen las predicciones. Especialistas como Lupas preparan estas tareas objetivo y evalúan los resultados cada dos años.
El nivel de acuerdo se evalúa regularmente utilizando una métrica llamada Prueba de distancia global (GDT, específicamente la «TS» o variante de puntuación total), que es el porcentaje de aminoácidos cuyas posiciones predichas están más cerca que algún umbral del valor experimental. Durante una década, los participantes de CASP han luchado para superar una puntuación media de 40 en la desafiante categoría de modelado libre.
DeepMind ingresó por primera vez a CASP en 2018, y su herramienta AlphaFold logró un GDT significativamente mejorado de 60. Para la evaluación de 2020, modificaron completamente su arquitectura para usar métodos basados en la atención, que se han destacado en modelos de lenguaje a gran escala. Este enfoque, que se basa en detalles de bajo nivel, difiere de la mayoría de los enfoques anteriores que comenzaron con una vista de arriba hacia abajo de la estructura completa. El equipo también utilizó el entrenamiento de extremo a extremo del modelo, reforzando las buenas predicciones de la estructura de las secuencias iniciales, en lugar de monitorear los pasos intermedios. Para la formación, utilizaron bases de datos que incluían unas 170.000 combinaciones de estructura / secuencia de proteínas, así como otros datos.
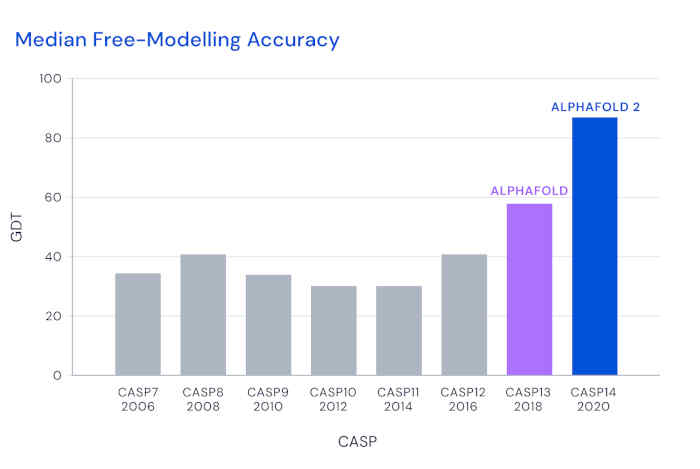
Con estos métodos, AlphaFold 2 logró un GDT de casi 90 en el último CASP, y cada estructura requirió solo unos pocos días de cálculo. Esta enorme mejora refleja una precisión aproximadamente comparable a la incertidumbre experimental. Lupas y sus colegas también «les preocupaba que les estuviera yendo demasiado bien», por lo que incluyeron una proteína para la que habían luchado por encontrar una estructura durante una década, a pesar de tener excelentes datos de rayos X. Dijo que «con su modelo, la estructura se derrumbó en unos pocos minutos».
DeepMind declinó una entrevista para esta historia, pero después de su éxito en 2018, la organización publicó muchos de los detalles técnicos. Aunque «no se han proporcionado suficientes detalles» hasta ahora, dijo Lupas, «AlphaFold ha sido bastante abierto con nosotros, diciéndonos cuáles fueron las ideas clave». Como resultado, «ahora todo el mundo está jugando con los nodos de atención. La mayoría de la gente está haciendo redes de extremo a extremo», dijo. «Hay muchos estudiantes de posgrado que no duermen mucho».
Poner en práctica la predicción
«A medida que se disponga de predicciones de estructuras cada vez más precisas, realmente ayudará a esclarecer la biología para la que antes habría necesitado una determinación estructural experimental», dijo David Baker de la Universidad de Washington, cuya herramienta Rosetta fue líder en evaluaciones CASP anteriores. Aún así, señaló, «por lo general, no es solo la estructura, sino también los experimentos en la parte superior de esa estructura, lo que se necesita para obtener conocimientos biológicos».
No obstante, AlphaFold y herramientas de predicción similares se pueden generalizar para iluminar la función biológica. Por ejemplo, las proteínas se unen frecuentemente entre sí en pares o en complejos más grandes, o al ADN o al ARN, para realizar funciones biológicas específicas. Las interacciones espaciales de proteínas con moléculas pequeñas también pueden mostrar cómo promueven reacciones químicas, o cómo esas pequeñas moléculas pueden servir como fármacos para modificar sus acciones.
Una cuestión pendiente, señaló Lupas, se refiere a las muchas proteínas, o regiones de proteínas, cuyas estructuras no están predeterminadas con precisión. «Todo en CASP tiene una estructura. Esa es una de las premisas», dijo. Espera saber si AlphaFold o sus sucesores pueden saber si una estructura en particular es única. «¿Esta red sabe cuando no hay solución?»
El equipo de Baker ha desarrollado principios para diseñar proteínas que no tienen análogos naturales, varios de los cuales se encuentran en ensayos clínicos. En un trabajo basado en la versión anterior de AlphaFold, «Realmente hizo un trabajo notablemente bueno al predecir la estructura de nuestro de novo proteínas diseñadas «, dijo Baker.» Estas redes claramente están aprendiendo principios generales «, pero no en una forma que la gente pueda entender fácilmente.
Don Monroe es un escritor de ciencia y tecnología con sede en Boston, MA, EE. UU.
entradas no encontradas