
Crédito: Verónica Falconieri Hays
Hace dos años, cuando la pandemia de COVID-19 se extendió por todo el mundo, los investigadores de DeepMind, la filial de inteligencia artificial (IA) y laboratorio de investigación de Alphabet Inc., demostraron cómo podría utilizar el aprendizaje automático para lograr un gran avance en la capacidad de predecir cómo las proteínas, los caballos de batalla de la célula viva, se pliegan en las formas intrincadas que toman. El trabajo dio esperanza a los biólogos de que podrían usar este tipo de herramienta para abordar enfermedades como el coronavirus SARS-CoV-2 mucho más rápido en el futuro.
Los investigadores pudieron evaluar las capacidades de AlphaFold2 de DeepMind gracias a su inclusión en el 14el Evaluación crítica de predicción de estructuras (CASP14), una competencia de evaluación comparativa que se desarrolló hasta 2020 y que agregó un programa paralelo para descubrir las estructuras de proteínas clave del virus SARS-CoV2 para intentar acelerar el desarrollo de vacunas y medicamentos. Los organizadores de CASP14 declararon que la herramienta representaba «una solución casi completa al problema de calcular la estructura tridimensional a partir de secuencias de aminoácidos», aunque hay algunas advertencias detrás de esa declaración.
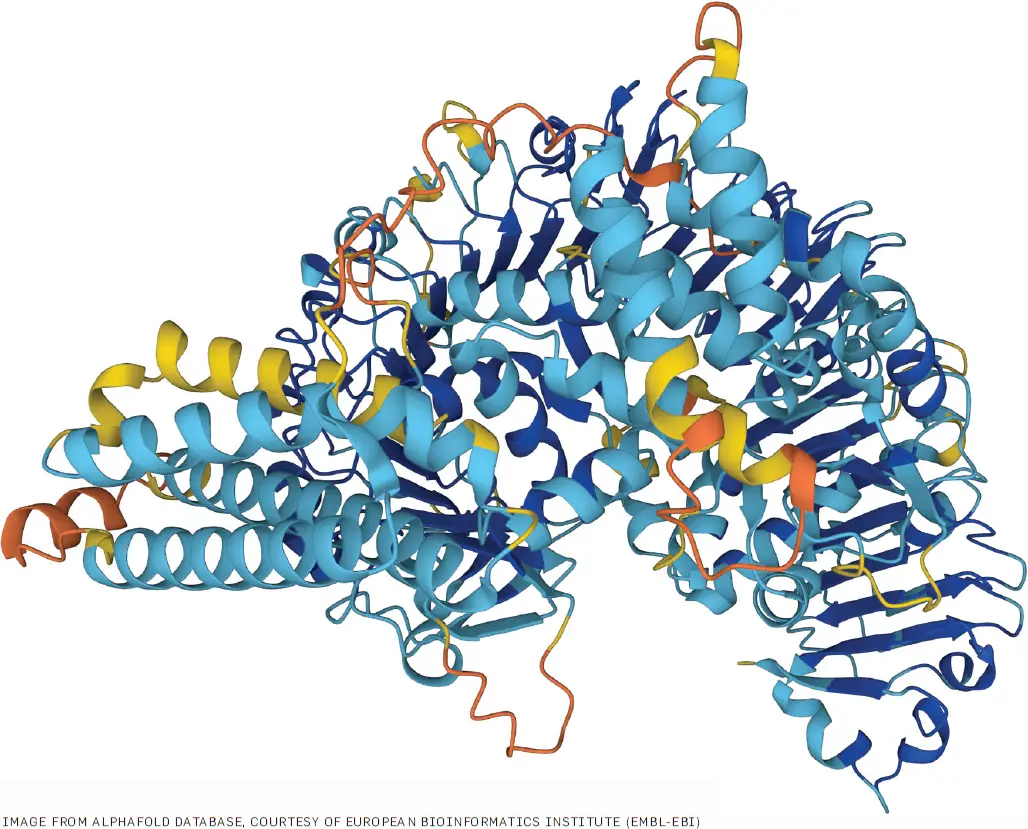
Cifra. Una predicción de proteína AlphaFold con una puntuación de confianza por residuo muy alta (superior a 90 de 100).
En principio, las simulaciones mecánicas cuánticas pueden predecir qué colección de pliegues conduce a la energía combinada más baja de todos los enlaces químicos en la forma y el agua y otras moléculas a su alrededor. Sin embargo, esto permanece más allá de la capacidad incluso de las computadoras actuales y puede que ni siquiera sea práctico en la mayoría de los casos.
John Jumper, científico investigador principal del personal de DeepMind, señala que realizar una simulación de dinámica molecular completa no solo es complejo desde el punto de vista computacional; requiere una especificación completa del entorno que rodea a la proteína en cuestión. «Las proteínas son máquinas exquisitamente sensibles y extremadamente finamente equilibradas. No podemos escribir funciones energéticas realmente buenas para ellas. Incluso pequeños cambios, como equivocarse en la concentración de sal o no especificar alguna condición, pueden hacer que no se plieguen en absoluto. Y no tienes ninguna esperanza de escribir todas las condiciones correctas de cada proteína en la célula humana», dice.
Cuando los biólogos producen estructuras para proteínas de forma experimental, encuentran formas de fijar la molécula en lo que esperan que sea una conformación representativa. Un método es aislar y cristalizar la proteína y luego usar la difracción de rayos X para estimar las posiciones de los átomos en la estructura compleja. Otro método cada vez más común es la microscopía electrónica criogénica (crio-EM): congelar la proteína aislada y luego usar la dispersión de haces de electrones por los átomos para determinar cómo se dobla y pliega la cadena de proteína. Años de esfuerzo han poblado bases de datos de acceso público, como el Banco de datos de proteínas (PDB), creado por un grupo de laboratorios del Reino Unido y los EE. UU. en 1971. Aunque su creación fue laboriosa, estos datos han demostrado ser cruciales para la creciente eficacia de los modelos basados en IA.
Algunos métodos de aprendizaje automático aplicados a principios de la década de 2010 se centraron en cómo evolucionan las secuencias de proteínas y su relación con las formas de PDB. Muchas proteínas poseen residuos de aminoácidos en posiciones clave que son importantes para determinar la estructura en las muchas variantes que se han acumulado en el registro evolutivo. Estos residuos suelen estar bastante separados en la cadena, pero se juntan en la versión plegada y mantienen la proteína en esa forma a través de enlaces formados dinámicamente por interacciones entre sus capas de electrones. Estas similitudes aparecen en las secuencias de aminoácidos que están disponibles para muchas proteínas: son mucho más baratas de obtener que la información sobre estructuras. Entrenar una red en estas similitudes, que se identifican mediante un proceso de alineación de secuencias múltiples (MSA), ayudó a reducir la cantidad de formas posibles diferentes que los motores de física debían considerar. Sin embargo, la precisión con el uso de estos modelos siguió siendo relativamente baja.
Eso comenzó a cambiar cuando los investigadores de aprendizaje automático vieron una conexión entre los datos almacenados en PDB y las redes neuronales convolucionales (CNN) utilizadas para analizar imágenes bidimensionales (2D). Una forma de representar los pliegues de las proteínas es utilizar un mapa de contactos. Esto coloca cada uno de los residuos de aminoácidos en la secuencia a lo largo de los ejes de una matriz, y se usa una puntuación en cada emparejamiento para representar qué tan cerca están en el espacio en el modelo PDB. RaptorX-Contact, desarrollado en el Instituto Tecnológico de Toyota en Chicago, obtuvo una alta calificación en el CASP12 de 2016 y utilizó una CNN basada en la arquitectura ResNet ampliamente utilizada, tratando de hecho el mapa de contacto como una entrada de imagen.
Las estimaciones de precisión probable proporcionadas por AlphaFold2 tienden a coincidir bien con los errores en la estructura generada.
AlphaFold2 se alejó de las CNN y cambió la arquitectura a una red neuronal basada en la atención entrenada en estructuras PDB en un sistema que también incorporó la técnica MSA utilizada en sistemas más antiguos. El resultado fue un modelo de 21 millones de parámetros lo suficientemente efectivo como para impulsar a AlphaFold2 por delante de sus competidores en CASP14.
Antes de que AlphaFold2 publicara el código del modelo, los investigadores del Baker Lab de la Universidad de Washington desarrollaron su propio enfoque para una red neuronal basada en la atención llamada RoseTTAFold. Al hacerlo, agregaron un tercer componente al modelo que considera las coordenadas tridimensionales (3D) de los residuos directamente para mejorar la precisión general. Sin embargo, AlphaFold2 entregó mejores resultados en promedio en CASP15, realizado a fines del año pasado.
Después de que el código fuente del modelo AlphaFold2 estuvo disponible, muchos equipos optaron por basar su propio trabajo en él y lo presentaron en CASP15, aunque DeepMind no participó directamente. El año pasado, un consorcio de laboratorios y empresas de tecnología lanzó OpenFold, una versión simplificada de AlphaFold2 desarrollada por Mohammed AlQuraishi, profesor asistente en el Departamento de Biología de Sistemas de la Universidad de Columbia y miembro del Programa de Genómica Matemática de Columbia. A diferencia del código disponible que ofrece DeepMind, OpenFold también incluye las herramientas para entrenar versiones personalizadas del modelo.
Los experimentadores también han aceptado los resultados de la IA, ayudados en parte por una base de datos alojada por el laboratorio paneuropeo de ciencias de la vida EMBL. Contiene estructuras predichas por AlphaFold2 para todas las proteínas humanas conocidas, muchas de las cuales aún no tienen entradas en PDB. Tom Terwilliger, científico principal del Consorcio de Nuevo México, dice que la ventaja de usar estructuras predichas por AlphaFold2 y herramientas similares en flujos de trabajo experimentales es que puede generar estructuras hipotéticas, a menudo cercanas a las formas físicas indicadas por los datos de crio-EM o rayos X, si no completamente exacto. En la práctica, las estimaciones de precisión probable proporcionadas por AlphaFold2 tienden a coincidir bien con los errores en la estructura generada.
Un caso de uso típico es tomar la estructura predicha y «acoplarla» a un mapa de densidad de electrones creado por mediciones crio-EM para determinar dónde difieren los dos y luego adaptar la estructura a una que se ajuste mejor al mapa de densidad. Esto se ha ampliado a un flujo de trabajo desarrollado por Terwilliger y sus colegas en el que luego se proporcionan refinamientos a AlphaFold2 para que pueda generar una nueva predicción que a menudo está mucho más cerca de los datos experimentales.
Algunos críticos argumentan que la falta de física en el modelo central representa un problema que los sistemas futuros deberán resolver. Si se realiza un pequeño cambio en la secuencia central dada a AlphaFold2, tal vez uno que interrumpa el acoplamiento entre dos de los residuos, el modelo puede no predecir el cambio en la estructura física.
Aun así, varios grupos han descubierto que pueden mejorar AlphaFold2 en el manejo de mutaciones sin cambiar su estructura central. Una técnica es usar cadenas mucho más cortas como entradas al proceso MSA. Para las proteínas con una buena representación en los datos existentes, estas estructuras a menudo se asemejan a las diferentes conformaciones que puede adoptar una proteína a medida que cambia el entorno que la rodea o cuando se unen y liberan otras moléculas e iones.
Algunos creen que el rendimiento de los pLM dependerá de la cantidad de datos utilizados para el entrenamiento y la cantidad de parámetros en el modelo.
«Hasta ahora, parece que simplemente dejar que el modelo aprenda todo, sin hacer suposiciones físicas previas, simplemente funciona mejor», dice AlQuraishi. «Sospecho que esto se debe en parte a que tenemos suficientes datos para entrenar un modelo de predicción de la estructura de proteínas, al menos si la arquitectura está bien diseñada, como la de AlphaFold2. Es concebible que para otros problemas moleculares donde los datos son más escasos, incluso para Por ejemplo, predecir los efectos de mutaciones estructuralmente disruptivas, incorporar la física dará sus frutos».
Los modelos de lenguaje grande como el BERT de Google pueden mejorar la capacidad de los sistemas basados en IA para predecir los efectos de las mutaciones, así como las formas probables de proteínas nuevas o inusuales donde las secuencias y estructuras relacionadas son escasas. Hasta ahora, los resultados de varios de los participantes del modelo de lenguaje de proteínas (pLM) no han alcanzado la precisión de AlphaFold2.
Al igual que con el procesamiento del lenguaje natural, algunos creen que el rendimiento de los pLM dependerá de la cantidad de datos utilizados para el entrenamiento y la cantidad de parámetros en el modelo, que aún no alcanzan los miles de millones que podrían ser necesarios. Konstantin Weissenow, un Ph.D. estudiante del Rost Lab de la Universidad Técnica de Munich en Alemania, dice que en el trabajo de ese equipo, los pLM no funcionan tan bien como se esperaba en proteínas que no tienen secuencias relacionadas, y los modelos más grandes no necesariamente funcionan mejor.
«Todavía se están investigando las razones. Pero podrían deberse al hecho de que el entrenamiento del modelo de lenguaje en sí mismo también está sesgado por la información disponible en las bases de datos de secuencias grandes. Espero que el diseño inteligente de las arquitecturas de aprendizaje automático resulte ser la parte más crítica en la mejora adicional de los sistemas de predicción de estructuras, basados o no en pLM», dice Weissenow.
En un número cada vez mayor de casos, los diferentes modelos de IA se utilizan en combinación. Un proyecto de Baker Lab intentó encontrar qué proteínas de levadura tienen más probabilidades de interactuar utilizando una forma simplificada de RoseTTAFold para filtrar 5500 emparejamientos de candidatos de un total de 4,3 millones. El AlphaFold2, más lento pero más preciso, redujo la lista más corta a 1.500 pares probablemente biológicamente activos. A principios de este año, la startup Evozyne dijo que había utilizado un pLM desarrollado junto con la empresa informática Nvidia para generar proteínas variantes para la síntesis rápidamente, con AlphaFold2 empleado para ayudar a confirmar visualmente las estructuras físicas.
Weissenow señala: «Personalmente, creo que los enfoques basados en pLM coexistirán con los métodos tradicionales basados en MSA durante al menos un tiempo. Si hay disponibles MSA ricos para una proteína, las predicciones de AF2 y sistemas similares serán la solución. para usuarios intermedios interesados en estructuras de la más alta calidad».
 Otras lecturas
Otras lecturas
AlQuraishi, M.
Aprendizaje automático en la predicción de la estructura de proteínas
Opinión actual en biología química 202165, 1–8
Jumper, J. et al.
Predicción altamente precisa de la estructura de proteínas con AlphaFold
Naturaleza 596, 583–589 (2021)
Terwilliger, TC et al.
Modelado AlphaFold mejorado utilizando información implícita de mapas de densidad experimentales
Métodos de la naturaleza 19, 1376-1382 (2022)
Weissenow, K., Heinzinger, M. y Rost, B.
Incorporaciones de modelos de lenguaje de proteínas para una predicción de estructura de proteínas rápida, precisa y sin alineación
Estructura30, 8, 1169–1177 (2022)
Volver arriba
Autor
Chris Edwards es un escritor residente en Surrey, Reino Unido, que informa sobre electrónica, TI y biología sintética.
©2023 ACM 0001-0782/23/05
Se otorga permiso para hacer copias digitales o impresas de parte o la totalidad de este trabajo para uso personal o en el aula sin cargo, siempre que las copias no se hagan o distribuyan con fines de lucro o ventaja comercial y que las copias lleven este aviso y la cita completa en la primera página. Deben respetarse los derechos de autor de los componentes de este trabajo que no pertenezcan a ACM. Se permite hacer resúmenes con crédito. Para copiar de otro modo, volver a publicar, publicar en servidores o redistribuir a listas, se requiere un permiso y/o tarifa específicos previos. Solicite permiso para publicar a permisos@acm.org o envíe un fax al (212) 869-0481.
La Biblioteca digital es una publicación de la Association for Computing Machinery. Derechos de autor © 2023 ACM, Inc.
entradas no encontradas
